













 Marcy's Musings: The Growing Industry
Marcy's Musings: The Growing Industry It’s Only Common Sense: Here’s What To Do After IPC APEX EXPO 2024
It’s Only Common Sense: Here’s What To Do After IPC APEX EXPO 2024 Dan’s Biz Bookshelf: Seeing the How
Dan’s Biz Bookshelf: Seeing the HowTesting New Drugs with "ALS-on-a-Chip"
October 11, 2018 | MITEstimated reading time: 4 minutes
There is no cure for amyotrophic lateral sclerosis (ALS), a disease that gradually kills off the motor neurons that control muscles and is diagnosed in nearly 6,000 people per year in the United States.
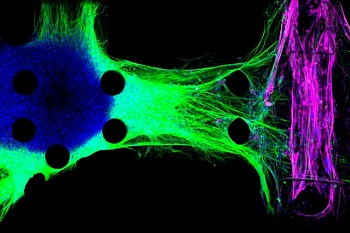
Image Caption: MIT engineers created this model of the neuromuscular junction using motor neurons derived from ALS patients. The motor neurons (blue) send fibers called neurites (green) toward the muscle fibers (pink). Credit: Tatsuya Osaki/MIT
In an advance that could help scientists develop and test new drugs, MIT engineers have designed a microfluidic chip in which they produced the first 3-D human tissue model of the interface between motor neurons and muscle fibers. The researchers used cells from either healthy subjects or ALS patients to generate the neurons in the model, allowing them to test the effectiveness of potential drugs.
“We found striking differences between the healthy cells and the ALS cells, and we’ve been able to show the effects of two drugs that are in clinical trials right now,” says Roger Kamm, the Cecil and Ida Green Distinguished Professor of Mechanical and Biological Engineering at MIT and the senior author of the study.
MIT postdoc Tatsuya Osaki is the lead author of the paper, which appears in the Oct. 10 issue of Science Advances. Sebastien Uzel, a former MIT graduate student, is also an author of the paper.
3-D Junctions
Scientists began developing tissue models of the connections between motor neurons and muscle cells, also called neuromuscular junctions, decades ago. However, these were limited to two-dimensional structures, which do not fully replicate the complex physiology of the tissue.
Kamm and his colleagues developed the first version of their 3-D neuromuscular junction model two years ago. The model consists of neurons and muscle fibers that occupy adjacent compartments of a microfluidic chip. Once placed in the compartments, the neurons extend long fibers called neurites, which eventually attach to the muscles, allowing the neurons to control their movement.
The neurons are engineered so that the researchers can control their activity with light, using a technique called optogenetics. The muscle fibers are wrapped around two flexible pillars, so when the neurons are activated by light, the researchers can measure how much the muscle fibers contract by measuring the displacement of the pillars.
In the 2016 version of the model, the researchers used mouse cells to grow the neurons and muscles, but differences between species can affect drug screening. In the new study, they used induced pluripotent stem cells from humans to generate both the muscle cells and the neurons. After demonstrating that the system worked, they began to incorporate neurons generated from induced pluripotent stem cells from a patient with sporadic ALS, which accounts for 90 percent of all cases.
This ALS model showed significant differences from the neuromuscular junctions created from healthy cells. The neurites grew more slowly and seemed to be unable to form strong connections with the muscle fibers, Kamm says.
“You can see that the healthy neurites are going directly to the individual myotubes and then activating them. However, the ALS neurons don’t seem to be able to connect very well,” he says.
This translated to weaker muscle control: After two weeks, the muscles innervated by ALS motor neurons were generating only about one-quarter the force produced by muscles controlled by healthy neurons. This also suggested that ALS motor neurons attacked healthy skeletal muscle tissues.
“The use of human-derived neuronal cells from ALS patients, combined with stem cell derived muscle cells — and the formation of a functional neuromuscular junction — is a major advance in the field of tissue models on a chip,” says Rashid Bashir, a professor of electrical and computer engineering and bioengineering at the University of Illinois at Urbana-Champaign, who was not involved in the research.
Promising Drugs
The researchers then used their model to test two drugs that are now in clinical trials to treat ALS — rapamycin and bosutinib. They found that giving both of the drugs together restored most of the muscle strength that had been lost in the ALS motor units. The treatment also reduced the rate of cell death normally seen in the ALS motor unit.
Working with a local biotech company, Kamm and his colleagues hope to collect induced pluripotent stem cells from 1,000 ALS patients, allowing them to perform larger-scale drug studies. They also plan to scale up the technology so they can test more samples at a time, and to add more types of cells, such as Schwann cells and microglial cells, which play supportive roles in the nervous system.
This tissue model could also be used to study other muscular diseases such as spinal muscular atrophy, which affects nerve cells found in the spine.
The research was funded by the National Science Foundation through the Science and Technology Center on Emergent Behaviors of Integrated Cellular Systems.
Share on:




Suggested Items
Connect the Dots: Best Practices for Prototyping
09/21/2023 | Matt Stevenson -- Column: Connect the DotsPCB prototyping is a critical juncture during an electronic device’s journey from concept to reality. Regardless of a project’s complexity, the process of transforming a design into a working board is often enlightening in terms of how a design can be improved before a PCB is ready for full production.
The Drive Toward UHDI and Substrates
09/20/2023 | I-Connect007 Editorial TeamPanasonic’s Darren Hitchcock spoke with the I-Connect007 Editorial Team on the complexities of moving toward ultra HDI manufacturing. As we learn in this conversation, the number of shifting constraints relative to traditional PCB fabrication is quite large and can sometimes conflict with each other.
Asia/Pacific AI Spending Surge to Reach a Projected $78 Billion by 2027
09/19/2023 | IDCAsia/Pacific spending on Artificial Intelligence (AI) ), including software, services, and hardware for AI-centric systems will grow to $78.4 billion in 2027, according to International Data Corporation's latest Worldwide Artificial Intelligence Spending Guide.
Intel to Sell Minority Stake in IMS Nanofabrication Business to TSMC
09/13/2023 | IntelIntel Corporation announced that it has agreed to sell an approximately 10% stake in the IMS Nanofabrication business to TSMC. TSMC’s investment values IMS at approximately $4.3 billion, consistent with the valuation of the recent stake sale to Bain Capital Special Situations.
RAF Invests in BAE Systems’ Most Advanced Fighter Pilot Helmet
09/13/2023 | BAE SystemsThe UK Ministry of Defence (MOD) has awarded BAE Systems a contract to develop its Striker II Helmet Mounted Display (HMD) for the Royal Air Force (RAF) Typhoon fleet. The contract, valued at £40m, will create and sustain more than 200 highly-skilled jobs at BAE Systems’ sites in Kent and Lancashire working directly on the Striker II programme. In total, the Typhoon programme sustains more than 20,800 jobs across the UK.